

NEXTFLEX® ChIP-Seq Library Prep Kit for Illumina® Sequencing
Optimized for down to 1 ng of DNA input
For use with ChIP or genomic DNA samples
Enhanced Adapter Ligation Technology offers optimal coverage and unique reads
- Product description
- Protocol
- Data
- Kit Contents
- Citations
Designed for Ultra-low DNA Input
The NEXTFLEX® ChIP-Seq Kit is a library prep kit specifically designed for ultra-low DNA input Illumina® sequencing applications. As little as 1 ng of chromatin immunoprecipitated or genomic DNA is required to produce viable sequencing libraries compatible with single-read or paired-end sequencing.
The NEXTFLEX® ChIP-Seq Kit also features proprietary Enhanced Adapter Ligation Technology, resulting in library preps with a larger number of unique sequencing reads and increased yields from minimal quantities of starting material. The NEXTFLEX® ChIP-Seq Kit now incorporates a bead-based size selection protocol, which completely eliminates the gel purification step. Additionally, there is an optional protocol which eliminates the size selection step entirely.
Unparalleled Multiplexing Capabilities
Using the NEXTFLEX® ChIP-Seq Barcodes, the user can index 6, 12, 24, 48, or even 96 samples at once providing unparalleled Illumina® ChIP sequencing capacity.
Features
Optimized for down to 1 ng of DNA input
For use with ChIP or genomic DNA samples
Optimized for low DNA input
Enhanced Adapter Ligation Technology offers optimal coverage and unique reads
Flexible barcode options – up to 96 unique, single-index barcoded adapters or 1,536 UDI barcodes available
Compatible with the Illumina® MiSeq®, HiSeq®, HiSeq®, HiSeq® X Ten, and NextSeq® 500 sequencing platforms
Kit Specs
| Cat # | Name | quantity |
| NOVA-5143-01 | NEXTFLEX® ChIP-Seq Library Prep Kit for Illumina® Sequencing | 8 RXNS |
| NOVA-5143-02 | NEXTFLEX® ChIP-Seq Library Prep Kit for Illumina® Sequencing | 48 RXNS |
| NOVA-514120 | COMPATIBLE ChIP-SEQ BARCODES 6 BARCODES | 48 RXNS |
| NOVA-514121 | COMPATIBLE ChIP-SEQ BARCODES 12 BARCODES | 96 RXNS |
| NOVA-514122 | COMPATIBLE ChIP-SEQ BARCODES 24 BARCODES | 192 RXNS |
| NOVA-514123 | COMPATIBLE ChIP-SEQ BARCODES 48 BARCODES | 384 RXNS |
Protocol
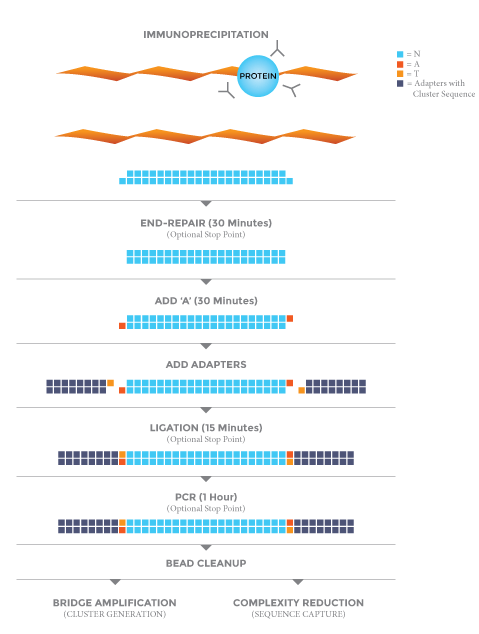
ACHIEVING HIGH COVERAGE & YIELD FROM GC & AT RICH GENOMES
Standard Next Generation Sequencing (NGS) library preparation methods include a polymerase chain reaction (PCR) amplification step prior to cluster generation and sequencing, to meet minimum molarity cutoffs required by different sequencing platforms. Biases associated with PCR amplification include uneven coverage of regions with extreme base composition, increased numbers of duplicate fragments, decreased mapping quality and poor variant calling. Particularly intractable regions of extreme base composition include GC-rich regions, which remain difficult to uniformly PCR amplify. While the human genome is not considered GC-rich (3.2 Gb; 41% GC), certain regions of the genome can be extremely GC rich, e.g. retinoblastoma tumor suppressor gene RB1 (104 of the first 136 coding bases are either G or C). Unbiased amplification of highly complex whole genome sequencing (WGS) libraries, whole exome sequencing (WES) libraries and targeted re-sequencing libraries is required to obtain high quality data for de novo genome assembly and accurate calling of single nucleotide polymorphisms (SNPs). Here, we show that the NEXTflex™ PCR polymerase is a leader in providing high quality amplification. The NEXTflex PCR polymerase, used in all of the NEXTflex DNA-seq and RNA-seq NGS library preparation kits, contains a highly optimized and robust enzyme that exhibits minimal GC bias and produces uniform coverage of difficult to sequence genomes.
Methods
Genomic DNA was isolated from Deinococcus radiodurans (3.2 Mb; 67% GC) and Saccharomyces cerevisiae (12.1 Mb; 38% GC). Genomic DNA was Covaris sheared to an average fragment size of 200 bp. 20 ng of D. radiodurans (GC-rich) and S. cerevisiae (AT-rich) DNA was used as starting material for library preparation using the NEXTflex™ ChIP-Seq Kit, following the gel-free protocol. Following barcoded-adapter ligation, GC-rich and AT-rich DNA samples were divided equally and independently PCR amplified for 14 cycles according to manufacturer’s instructions. Each library was prepared in duplicate. Average library size was assessed by Bioanalyzer® and concentrations determined by qPCR. Normalized libraries were clustered on-board, and 150 bp paired-end sequencing was performed on the HiSeq® 2500 across 4 lanes of 2 flowcells using rapid run mode.
11 different PCR enzymes used in library preparation kits for next generation sequencing were tested in this sequencing experiment. The performance of the three best are highlighted here: NEXTflex PCR polymerase, Supplier D and Supplier I. This subset of enzymes produced the most consistent metrics, including library yield, GC bias and raw coverage, of the 11 PCR kits tested across two genomes. Quality control sequencing metrics against the AT-rich genome show more paired-end reads for the NEXTflex PCR polymerase, and more total alignments and non-duplicated fragments than Suppliers D and I (Table 1).
RESULTS
Library Yield and Complexity
Several metrics, including library yield, were utilized to compare polymerase performance. Achieving sufficient library yields is important in order to meet minimum molarity cutoffs required by different sequencing platforms. Each polymerase produces distinct library quantities (Figure 1). Library yield comparison across the AT-rich genome shows a similar trend: libraries PCR amplified using NEXTflex PCR polymerase or Supplier D’s enzyme produce almost two times more library product than Supplier I. Conversely, across the GC-rich genome, NEXTflex PCR polymerase and Supplier I are equal; however, Supplier D library yields are reduced. This result shows the robust ability of the NEXTflex PCR polymerase to equally amplify both GC-rich and AT-rich genomes, and the inconsistent PCR efficiency of Suppliers D and I when using the same template for PCR. Library yields are important, but can give rise to misleading interpretations. Library yield inconsistencies produced by Supplier D suggest an unequal amount of AT-rich over GC-rich clones. Although library yields for Supplier D over the AT-rich genome are roughly equal to library yields of NEXTflex PCR polymerase (Figure 1), sequence complexity and richness (Table 1) are lower, while GC bias is higher and coverage is uneven (Figures 2, 3 and 4). The same trend is evident for Supplier I (Figure 2, 3 and 4).
GC Bias
To obtain a global representation of each PCR enzyme’s effect on sequence integrity, we examined PCR-introduced GC bias across both S. cerevisiae and D. radiodurans genomes using Picard GC bias plots (Figure 2). Low GC bias would produce a normalized coverage slope of one, or a horizontal line over windows of GC-rich content. Alternatively, high GC bias would produce a normalized coverage slope greater than one, over windows of GC-rich content (Figure 2, S. cerevisiae, supplier I). Picard analysis shows NEXTflex PCR polymerase to be the most consistently unbiased polymerase tested, regardless of genomic complexity and extreme base composition. Across the S. cerevisiae genome, NEXTflex PCR polymerase showed the lowest level of GC bias, with close to mean coverage across the entire genome, including GC-rich and AT-rich stretches. Unlike the NEXTflex PCR polymerase, normalized coverage of Supplier I across the S. cerevisiae genome is noticeably biased and uneven, producing uneven coverage of the genome, with fewer reads mapping to GC-rich regions and higher levels of PCR duplication in AT-rich regions. Conversely, the most average normalized coverage across the D. radiodurans genome was produced by Supplier I. NEXTflex PCR polymerase and Supplier D had slightly higher GC bias over GC-rich windows of the genome. Overall, however the NEXTflex PCR polymerase provides a balanced representation of both AT-rich and GC-rich regions, which neither Polymerase D or I attains.
Genome Coverage
In addition to GC bias, coverage across average GC-rich and AT-rich regions was considered for each polymerase. Across all four chromosomes of the D. radiodurans genome, NEXTflex PCR polymerase consistently outperforms Suppliers D and I, with higher levels of unbiased coverage (Figure 3). The NEXTflex PCR polymerase produced 110-160x coverage of the entire D. radiodurans genome compared to 75-110x coverage and 95-130x coverage for Suppliers D and I, respectively. Similar to coverage data of the GC-rich genome, the NEXTflex PCR polymerase shows higher coverage of the first eight chromosomes of the AT-rich genome than both Suppliers D and I (Figure 4; coverage of chromosomes 1-8 shown, 9-16 showed similar results). The NEXTflex PCR polymerase produced 30-40x coverage of the first eight chromosomes of the S. cerevisiae genome, compared to 25-35x coverage and 20-25x coverage for Suppliers D and I, respectively.
CONCLUSIONS
Our comparison between the NEXTflex PCR polymerase and those of Suppliers D and I found the NEXTflex PCR polymerase, which is used in all of the NEXTflex DNA library prep kits, including the NEXTflex ChIP-Seq Kit, and RNA-seq NGS library preparation kits, to be the most unbiased enzyme for NGS library prep across two highly diverse genomes, while consistently delivering even and high read coverage. Consistency of library yield between these two genomes demonstrates that this enzyme is extremely robust and can be applied to numerous NGS applications including, but not limited to, WGS, WES, metagenomics, target sequencing and other uses.
KIT CONTENTS
NEXTFLEX® ChIP End Repair Buffer Mix
NEXTFLEX® ChIP End Repair Enzyme Mix
NEXTFLEX® ChIP Adenylation Mix
NEXTFLEX® ChIP Ligation Mix
NEXTFLEX® ChIP Adapter (0.6 µM)
NEXTFLEX® ChIP Primer Mix (12.5 µM)
NEXTFLEX® ChIP PCR Master Mix
Nuclease-free Water
Resuspension Buffer
ChIP-Seq DNA Control
REQUIRED MATERIALS NOT PROVIDED
1 ng – 10 ng of DNA in up to 40 µL nuclease-free water
NEXTFLEX® ChIP-Seq Barcodes – 6 / 12 / 24 / 48
Ethanol 100% (room temperature)
Ethanol 80% (room temperature)
Covaris System (S2, E210) or other device for fragmenting DNA
96 well PCR Plate Non-skirted (Phenix Research®, Cat # MPS-499) / or / similar
96 well Library Storage and Pooling Plate (Fisher Scientific®, Cat # AB-0765) / or / similar
Adhesive PCR Plate Seal (Bio-Rad®, Cat # MSB1001)
Agencourt® AMPure® XP 5 mL (Beckman Coulter Genomics®, Cat # A63880)
Magnetic Stand -96 (Thermo Fisher® Scientific, Cat # AM10027) / or / similar
Heat block
Thermocycler
2, 10, 20, 200 and 1000 µL pipettes / multichannel pipettes
Nuclease-free barrier pipette tips
Microcentrifuge
1.5 mL nuclease-free microcentrifuge tubes
Vortex
Selected Publications that Cite Using the NEXTFLEX® ChIP-Seq Kit:
Achour, M. et al. (2015) Neuronal Identity Genes Regulated by Super-Enhancers Are Preferentially Down-Regulated in the Striatum of Huntington’s Disease Mice. Hum. Mol. Genet. 2015 : ddv099v1-ddv099.
Auclair, G., et al. (2015) EHMT2 directs DNA methylation for efficient gene silencing in mouse embryos. Genome Res. 26: 192-202. doi: 10.1101/gr.198291.115.
Baker, C. L., et al. (2015) Multimer Formation Explains Allelic Suppression of PRDM9 Recombination Hotspots. PLOS Genetics. doi: 10.1371/journal.pgen.1005512.
Baker, C. L., et al. (2015) PRDM9 Drives Evolutionary Erosion of Hotspots in Mus musculus through Haplotype-Specific Initiation of Meiotic Recombination. PLOS Genetics. doi: 10.1371/journal.pgen.1004916.
Baker, C. L., et al. (2014) PRDM9 binding organizes hotspot nucleosomes and limits Holliday junction migration. Genome Res. 24(5):724-32.
Bjorn-Mortensen, K., et al. (2015) Direct DNA Extraction from Mycobacterium tuberculosis Frozen Stocks as a Reculture-Independent Approach to Whole-Genome Sequencing. J. Clin. Microbiol. 53:8 2716-2719. doi:10.1128/JCM.00662-15.
Boo, K., et al. (2015) Pontin functions as an essential coactivator for Oct4-dependent lincRNA expression in mouse embryonic stem cells. Nature Communications 6:6810. doi:10.1038/ncomms7810.
Bubier, J. et al. (2014) Identification of a QTL in Mus musculus for Alcohol Preference, Withdrawal, and Ap3m2 Expression Using Integrative Functional Genomics and Precision Genetics. Genetics 197(4): 1377-1393.Chiu, W. T. et. al. (2014) Genome-wide view of TGFβ/Foxh1 regulation of the early mesendoderm program. Development. 141:4537-4547.
Clop A, Bertoni A, Spain SL, Simpson MA, Pullabhatla V, et al. (2013) An In-Depth Characterization of the Major Psoriasis Susceptibility Locus Identifies Candidate Susceptibility Alleles within an HLA-C Enhancer Element. PLoS ONE 8(8): e71690. doi:10.1371/journal.pone.0071690
Cvejic A, Haer-Wigman L, Stephens JC et al. (2013) SMIM1 underlies the Vel blood group and influences red blood cell traits. Nature Genetics. doi:10.1038/ng.2603
Deng, X. et al. (2014) Molecular mechanisms of two-component system RhpRS regulating type III secretion system in Pseudomonas syringae. Nucl. Acids Res. 42 (18): 11472-11486 doi: 10.1093/nar/gku865
Derboven, E., Ekker, H., Kusenda, B., Bulankova, P., Riha K. (2014) Role of STN1 and DNA Polymerase α in Telomere Stability and Genome-Wide Replication in Arabidopsis. PLoS Genetics. DOI: 10.1371/journal.pgen.1004682.
De Nardo, D, et. al. (2014) High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3 Nature Immunology 15:152-60.Dong, Q., Fang, M., Roychowdhury, S. and Bauer, C. E. (2015) Mapping the CgrA regulon of Rhodospirillum centenum reveals a hierarchal network controlling Gram-negative cyst development. BMC Genomics. 16:1066. doi:10.1186/s12864-015-2248-z.
Filleton, F., Chuffart, F., Nagarajan, M., Bottin-Duplus, H. and Yvert, G. (2015) The complex pattern of epigenomic variation between natural yeast strains at single-nucleosome resolution. Epigenetics Chromatin. 8:26. doi: 10.1186/s13072-015-0019-3.
Fischer, N., et al. (2014) Rapid metagenomic diagnostics for suspected outbreak of severe pneumonia [letter]. Emerg Infect Dis. doi: 10.3201/eid2006.131526.
Garrido, D., et al. (2015) Comparative transcriptomics reveals key differences in the response to milk oligosaccharides of infant gut-associated bifidobacteria. Scientific Reports. 5:13517. doi:10.1038/srep13517.
Golomb, B. L., Hirao, L. A., Dandekar, S. and Marco, M. L. (2016) Gene expression of Lactobacillus plantarum and the commensal microbiota in the ileum of healthy and early SIV-infected rhesus macaques. Scientific Reports 6:24723. doi:10.1038/srep24723.
Gosselin, D. et al. (2014) Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell. 159:6, 1327–1340. doi:10.1016/j.cell.2014.11.023.
Gregor, A. et al. (2013) De Novo Mutations in the Genome Organizer CTCF Cause Intellectual Disability. AJHG. 93:1. 124–131.
Jiang, L. et al. (2014) ZBED6 Modulates the Transcription of Myogenic Genes in Mouse Myoblast Cells. PLoS ONE 9(4): e94187. doi: 10.1371/journal.pone.0094187.
Jones, CJ. et. al. (2014) ChIP-Seq and RNA-Seq reveal an AmrZ-mediated mechanism for cyclic di-GMP synthesis and biofilm development by Pseudomonas aeruginosa. PLoS Pathog. e1003984.
Juntawong, P., Girke, T., Bazin, J. and Bailey-Serres, J. (2014) Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 111:1, E203-12.
Kieffer-Kwon, KR, et. al. (2013) Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell 155:1507-20.
Kong, W., et al. (2015) ChIP-seq reveals the global regulator AlgR mediating cyclic di-GMP synthesis in Pseudomonas aeruginosa. Nucl. Acids Res. 43:17. 8268-8282, doi: 10.1093/nar/gkv747.
Le Billan, F., et al. (2015) Cistrome of the aldosterone-activated mineralocorticoid receptor in human renal cells. FASEB J. 29: 3977 – 3989.
Lelandais, G., Blugeon, C. and Merhej, J. (2016) ChIPseq in Yeast Species: From Chromatin Immunoprecipitation to High-Throughput Sequencing and Bioinformatics Data Analyses. Yeast Functional Genomics: Methods and Protocols, Methods in Molecular Biology, 1361. Pg 1-11. Springer Science+Business Media New York. Doi: 10.1007/978-1-4939-3079.
Liang, H., et. al. (2014) Molecular mechanisms of master regulator VqsM mediating quorum-sensing and antibiotic resistance in Pseudomonas aeruginosa. Nucleic Acids Research. doi: 10.1093/nar/gku586.
Lam MTY, Cho H, Lesch HP et al. (2013) Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. doi:10.1038/nature12209.
Malysheva, V., Mendoza-Parra, M. A., Saleem, M.-A. M. and Gronemeyer, H. (2016) Reconstruction of gene regulatory networks reveals chromatin remodelers and key transcription factors in tumorigenesis. Genome Medicine. 8:57. doi: 10.1186/s13073-016-0310-3.
Mendoza-Parra, M.-A., et al. (2016) Antibody performance in ChIP-sequencing assays: From quality scores of public data sets to quantitative certification. F1000 Research. doi: 10.12688/f1000research.7637.1v1.
Parobek, C. M., Bailey, J. A., Hathaway, N. J., Socheat, D., Rogers, W. O. and Juliano, J. J. (2014) Differing Patterns of Selection and Geospatial Genetic Diversity within Two Leading Plasmodium vivax Candidate Vaccine Antigens. PLoS Neglected Tropical Diseases. doi: 1 0.1371/journal.pntd.0002796.
Pfeiffer, A., Shia, H., Teppermana, J. M., Zhanga, Y. and Quaila, P. H. (2014) Combinatorial Complexity in a Transcriptionally Centered Signaling Hub in Arabidopsis. Mol. Plant 7 (11): 1598-1618. doi: 10.1093/mp/ssu087.
Proudhon, C., et al. (206) 1Active and Inactive Enhancers Cooperate to Exert Localized and Long-Range Control of Gene Regulation. Cell Reports. 15:10, 2159–2169. doi: 10.1016/j.celrep.2016.04.087.
Rosario, R. C. H., et. al. (2015) Sensitive detection of chromatin-altering polymorphisms reveals autoimmune disease mechanisms. Nature Methods. doi:10.1038/nmeth.3326.
Rossetto CC, Tarrant-Elorza M, Pari GS (2013) Cis and Trans Acting Factors Involved in Human Cytomegalovirus Experimental and Natural Latent Infection of CD14 (+) Monocytes and CD34 (+) Cells. PLoS Pathog 9(5): e1003366. doi:10.1371/journal.ppat.1003366.
Rube, H. T., et al. (2016) Sequence features accurately predict genome-wide MeCP2 binding in vivo. Nature Communications. 7:11025. doi:10.1038/ncomms11025.
Sakashita, A., et al. (2015) Sex Specification and Heterogeneity of Primordial Germ Cells in Mice. PLoS ONE. 10:12. doi:10.1371/journal.pone.0144836.
Salehi, F., et al. (2015) CHOPER Filters Enable Rare Mutation Detection in Complex Mutagenesis Populations by Next-Generation Sequencing. PLoS ONE. Doi: 10.1371/journal.pone.0116877.
Scharer, C. D., et al. (2015) Genome-wide CIITA-binding profile identifies sequence preferences that dictate function versus recruitment. Nuc. Acids Res. doi: 10.1093/nar/gkv182.
Shin, H.-J., R., et al. (2016) AMPK–SKP2–CARM1 signalling cascade in transcriptional regulation of autophagy. Nature, doi:10.1038/nature18014.
Stelloh, C., et al. (2016) The cohesin-associated protein Wapal is required for proper Polycomb-mediated gene silencing. Epigenetics & Chromatin. 9:14, doi: 10.1186/s13072-016-0063-7.
Stenzig, J., et al. (2016) DNA methylation in an engineered heart tissue model of cardiac hypertrophy: common signatures and effects of DNA methylation inhibitors. Basic Research in Cardiology. 111:9. doi: 10.1007/s00395-015-0528-z.
Thakur J. and Sanyal K. (Feb 2013) Efficient neocentromere formation is suppressed by gene conversion to maintain centromere function at native physical chromosomal loci in Candida albicans. Genome Research. doi: 10.1101/gr.141614.11
Watson E., MacNeil L.T. et al. (2013) Integration of Metabolic and Gene Regulatory Networks Modulates the C. elegans Dietary Response. Cell.
Wu, Y, et al. (2014) Phosphorylation of p53 by TAF1 inactivates p53-dependent transcription in the DNA damage response. Mol. Cell 53(1):63-74.
Xiong, W., Li, J. Zhang, E. and Huang, H. (2016) BMAL1 regulates transcription initiation and activates circadian clock gene expression in mammals. Biochemical and Biophysical Research Communications. doi:10.1016/j.bbrc.2016.04.009.
Xu, Z., Chen, H., Ling, J., Yu, D., Struffi, P., and Small, S. (2014) Impacts of the ubiquitous factor Zelda on Bicoid-dependent DNA binding and transcription in Drosophila. Genes & Dev. 28: 608 – 621.
Xue, X., et al. (2015) LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene. doi:10.1038/onc.2015.340.
Yamane A., et al. (2013) RPA Accumulation during Class Switch Recombination Represents 5′–3′ DNA-End Resection during the S–G2/M Phase of the Cell Cycle. Cell Reports.
Yang, H., Kwon, C. S., Choi, Y. and Lee, D. (2016) Both H4K20 mono-methylation and H3K56 acetylation mark transcription-dependent histone turnover in fission yeast. Biochemical and Biophysical Research Communications. doi: 10.1016/j.bbrc.2016.05.155.
Zhao, J., et al. (2016) Structural and Molecular Mechanism of CdpR Involved in Quorum-Sensing and Bacterial Virulence in Pseudomonas aeruginosa. doi: 10.1371/journal.pbio.1002449.






